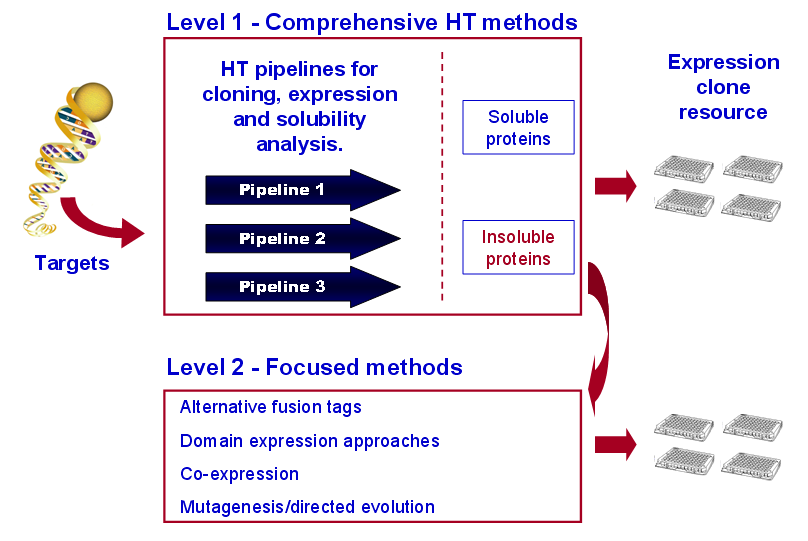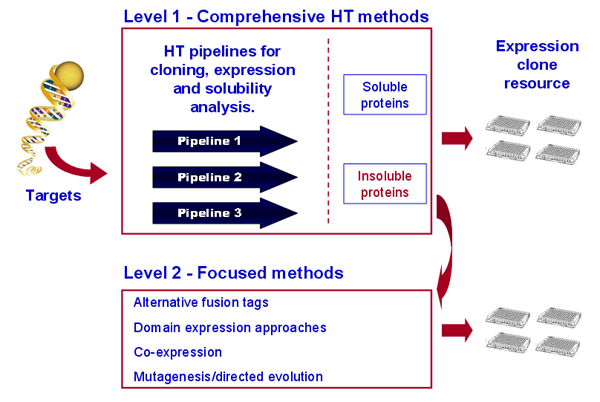
 To meet the production goals for PSI-2, we apply a comprehensive 96-well-plate HTP technology to generate clones and express soluble proteins. Our pipelines use pMCSG7 as the primary expression vector and a maltose-binding protein (MBP) fusion vector for a “salvage” strategy for proteins that express well in pMCSG7 but show low solubility. Expression clones that produce insoluble proteins are directed to Level 2 processing (see Figure). The developmental goal is to address solubility problems using HTP approaches. Criteria for entry into the salvage loop will include: lack of a soluble orthologues, poor diffraction quality crystals, or high target priority due to biomedical impact. This tiered strategy leverages our efficient and cost-effective parallel processes designed for mass production of proteins and protein fragments in E. coli. To meet the production goals for PSI-2, we apply a comprehensive 96-well-plate HTP technology to generate clones and express soluble proteins. Our pipelines use pMCSG7 as the primary expression vector and a maltose-binding protein (MBP) fusion vector for a “salvage” strategy for proteins that express well in pMCSG7 but show low solubility. Expression clones that produce insoluble proteins are directed to Level 2 processing (see Figure). The developmental goal is to address solubility problems using HTP approaches. Criteria for entry into the salvage loop will include: lack of a soluble orthologues, poor diffraction quality crystals, or high target priority due to biomedical impact. This tiered strategy leverages our efficient and cost-effective parallel processes designed for mass production of proteins and protein fragments in E. coli.
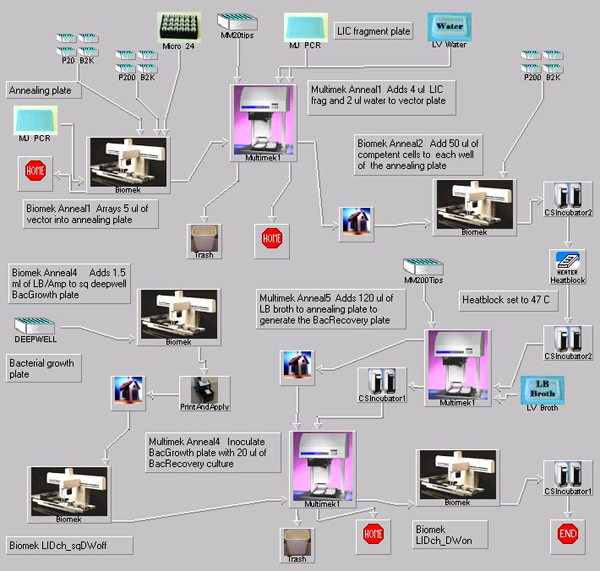
Coding regions are amplified using primers designed with the Express Primer tool or domain-specific primer design tools. All primers contain ligation-independent cloning sites compatible with multiple vectors. Affinity tags with a TEV (tobacco etch virus) protease cleavage site are fused to all proteins to facilitate their purification or capture. The primary steps of the process — PCR gene amplification, testing for protein expression and solubility — are conducted in 96-well-plate format. Denaturing PAGE analysis of proteins is carried out in a high-density gel format.
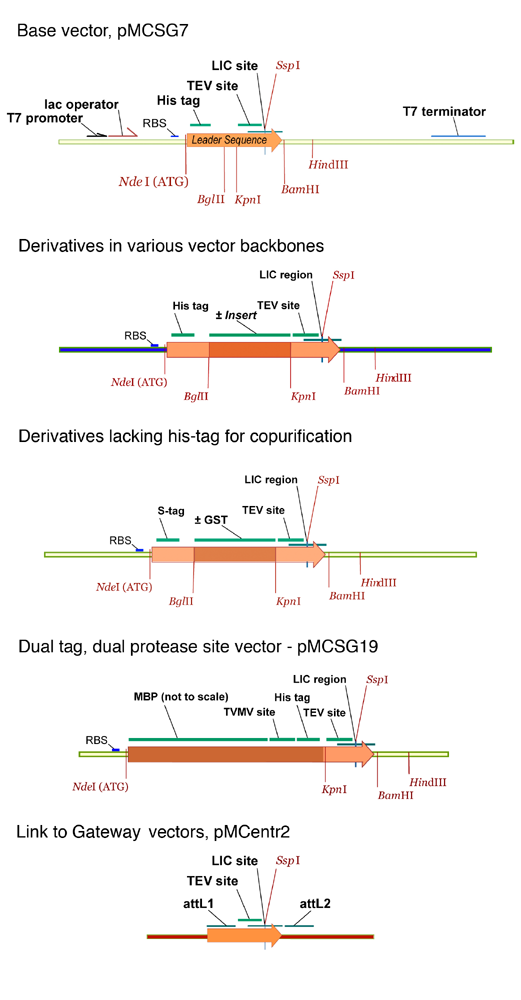
When the PSI pilot centers were formed, ligation-independent cloning (LIC) offered an attractive technology adaptable for robotic cloning, but existing vectors were not suitable for automated purification of proteins for crystallization. We developed a set of superior LIC vectors tailored specifically for this purpose. The vector, pMCSG7, encodes a His6-tag followed by a spacer and a TEV protease cleavage site that overlaps with the LIC site. This design puts the TEV site close to the start of the cloned native protein. Only the three-amino-acid-sequence SerAsnAla
When the PSI pilot centers were formed, ligation-independent cloning (LIC)
offered an attractive technology adaptable for robotic cloning, but existing
vectors were not suitable for automated purification of proteins for
crystallization. We developed a set of superior LIC vectors tailored
specifically for this purpose. The vector, pMCSG7, encodes a His6-tag followed
by a spacer and a TEV protease cleavage site that overlaps with the LIC site.
This design puts the TEV site close to the start of the cloned native protein.
Only the three-amino-acid-sequence SerAsnAla (SNA) is added to the protein after
protease cleavage.
For more information on vectors please see the vector
summary page.

TEV protease is highly specific and we have yet to observe substantial target
degradation. We also constructed a series of derivatives of pMCSG7 that fuse
helper peptides or proteins, such as MBP, to the N-termini of proteins or
introduce these elements into vectors with different origins of replication to
allow co-expression of proteins. Four additional vectors improve tandem
purification of complexes, aid robotic screening protocols, and improve robotic
protein purification. The pMCSG21 vector creates a bridge to Gateway
(Invitrogen) vectors to offer easy access to vectors designed to express
proteins in alternative hosts. As this avenue of protein production becomes more
important, some Gateway vectors will be redesigned to make them compatible with
the existing protein production pipelines. Gene expression in all the MCSG
vectors is driven by the T7 promoter and controlled by lac repressor, and all
vectors accept the same PCR products.
Vector |
Base Vector |
Encoded Leader Sequence |
Use |
pMCSG7 |
pET21a |
N-His-TEV-LICs- |
Routine protein production |
pMCSG8 |
pMCSG7 |
N-His-Sloop-TEV-LICs |
Improve solubility |
pMCSG9 |
pMCSG7 |
N-His-MBP-TEV-LICs |
Improve solubility |
pMCSG10 |
pMCSG7 |
N-His-GST-TEV-LICs |
Improve solubility |
pMCSG11 |
pACYCDuet-1 |
N-His-TEV-LICs |
Coexpression |
pMCSG12 |
pACYCDuet-1 |
N-His-Sloop-TEV-LICs |
Coexpression |
pMCSG13 |
pACYCDuet-1 |
N-His-MBP-TEV-LICs |
Coexpression |
pMCSG14 |
pACYCDuet-1 |
N-His-GST-TEV-LICs |
Coexpression |
pMCSG17 |
pMCSG7 |
N-Stag-TEV-LICs |
Coexpression |
pMCSG20 |
pMCSG7 |
N-Stag-GST-TEV-LICs |
Coexpression |
pMCSG16 |
pMCSG7 |
N-His-AviTag-TEV-LICs |
Phage display |
pMCSG15 |
pMCSG7 |
LICs-TEV-AviTag-His-C |
Phage display |
pMCSG18 |
pMCSG7 |
N-His-TEV-LICs-GFP |
Screening |
PMCSG19 |
pMCSG7 |
N-MBP-TVMV-His-TEV-LICs |
Purification |
pMCSG21 |
pDONR/zeo |
attL1-TEV-LIC-attL2 |
Gateway cloning |
For more information on vectors please see the vector summary page.
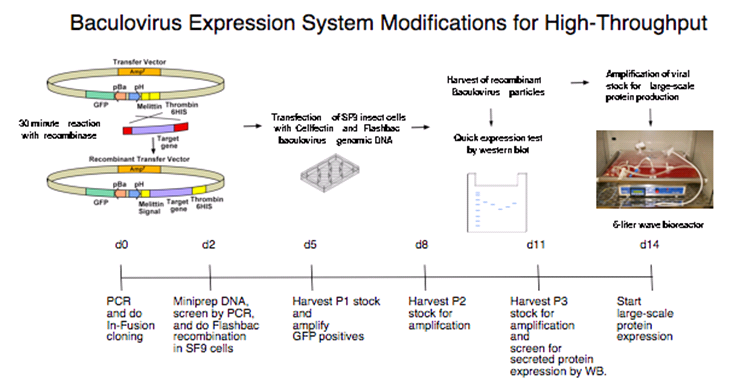
The insect cell expression system developed in the Fremont laboratory at Washington University is particularly well suited for targets that must be handled separately because they require correct disulfide bonds and other posttranslational modifications to produce properly folded proteins. The approach takes advantage of the fact that very few proteins are secreted from insect cells during baculovirus infection. Methods for the efficient recovery of secreted proteins from insect cell supernatants based on a His6 affinity tag have been developed. In addition, the fusion tag allows for easy monitoring of the infection and purification steps as it is easily detected on western blots using anti-His6 antiserum. To greatly shorten this process, the following modifications were implemented:
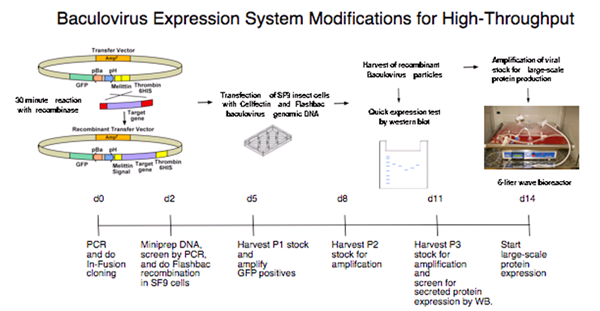
The transfer vector was modified to allow for ligation-independent cloning (LIC) of PCR fragments. The baculovirus transfer vector pAcUW51 was altered to contain a honeybee melittin signal sequence after the polyhedrin promoter. The honeybee melittin signal sequence has been shown to enhance the secretion of numerous foreign proteins from insect cells. Also, a C-terminal His6 tag removable by thrombin is included downstream of the cloning site.
We have succeeded in developing high-throughput bacterial inclusion body refolding protocols with particular emphasis on the folding of disulfide-bonded proteins. Again, for a typical target, we first PCR amplify the DNA corresponding to the mature secreted protein without the predicted leader sequence, transmembrane or intracellular regions, and then inserted it into a tagless pET-23b expression construct. For protein production we use BL21-Codon Plus (DE3)-RIL cells and induce expression with IPTG. Induced cell pellets are collected by centrifugation and lysed by sonication. Proteins are then recovered in the form of inclusion bodies and purified. The target proteins are first denatured, reduced, and then refolded by dilution under oxidative conditions. We have found small molecule additives like L-Arginine and NDSB to be extremely useful in optimizing refolding efficiencies. We next concentrate the refolded material and subject it to size exclusion chromatography. Further purification is usually pursued using ion-exchange chromatography, with protein identity and disulfide bond formation checked by mass spectrometry. For proteins with known ligands, we confirm correct folding of the recombinant reparations by testing their functional properties, for instance using surface plasmon resonance binding assays. For proteins where no known function exists, we judge appropriate folding by biophysical parameters that correlate with folding, including monodisperse profiles on size-exclusion chromatography and significant secondary structure as measured by circular dichroism spectroscopy.
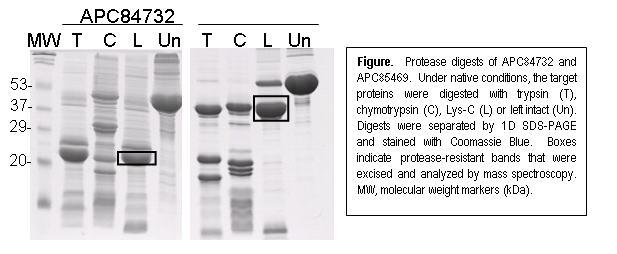
We are developing a salvage pathway for proteins that express well but fail in the crystallization trials. It is possible that crystallization of such proteins is inhibited by unfolded or disordered portions of the protein. Therefore, we are seeking to define the stable, folded domains of proteins through limited proteolysis. Target proteins are digested with various proteases under native conditions, and the protease-resistant portions of the protein that remain after digestion are analyzed by electrospray mass spectrometry to determine their intact mass and by tandem mass spectroscopy (ESI MS/MS) to determine their amino acid sequence. Bioinformatics is used to predict secondary structure and, together with data from the proteolysis experiments, guides the design of truncated constructs that can then be fed back through the cloning and crystallization pipeline.
1 MHHHHHHSSG VDLGTENLYF QSNAMKPIDR FSYLKNNRVS QDTSSLVQCY
51 LPIIGQEALS LYLYTISFWD NGRKEYLFSS ILNHLNFGMD RLIKSLKILS
101 AFNLLTLYQK GDVYQLALHA PLSSQDFLGH PVYRRLLEKK IGDVAVEDLK
151 VESADGEEIP VSLNQVFPEL AELGSQEDLG LKKKVANDFD LEHFRQLMAR
201 DGLRFADEQS DVLNLFAIAE EKKWTWFETY QLAKSTAVSQ VISTKRMREK
251 IAQKPVSSDF SLKEATIIKE AKSKTALQFL AEIKQTRKGT ITQTERELLQ
301 QMAGLGLLDE VINIILLLTF NKVDSANINE KYAMKVANDY AYQKIHSAEE
351 AVLRIRDRGQ KAKTQKQNQT APEKTNVPKW SNPEYKNETS EETRLELERK
401 KQELLARLEK G
 Selected related publications: Selected related publications:
Brett TJ, Legendre-Guillemin V, McPherson PS, Fremont DH (2006)
Structural definition of the F-actin-binding THATCH domain from HIP1R.
Nat Struct Mol Biol, 13, 121-30 Times cited: 6. [PubMed] [PDB]
Dieckman L, Gu M, Stols L, Donnelly MI, Collart FR (2002)
High throughput methods for gene cloning and expression.
Protein Expr Purif, 25, 1-7 Times cited: 42. [PubMed]
Donnelly MI, Stevens PW, Stols L, Su SX, Tollaksen S, Giometti C, Joachimiak A (2001)
Expression of a highly toxic protein, Bax, in Escherichia coli by attachment of a leader
peptide derived from the GroES cochaperone. Protein Expr Purif, 22, 422-9 Times cited: 7. [PubMed]
Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A (2006)
An expression vector tailored for large-scale, high-throughput purification of recombinant proteins.
Protein Expr Purif, 47, 446-54 Times cited: 5. [PubMed]
Moy S, Dieckman L, Schiffer M, Maltsev N, Yu GX, Collart FR (2004)
Genome-scale expression of proteins from Bacillus subtilis.
J Struct Funct Genomics, 5, 103-9 Times cited: no data. [PubMed]
Nelson CA, Pekosz A, Lee CA, Diamond MS, Fremont DH (2005)
Structure and intracellular targeting of the SARS-coronavirus Orf7a accessory protein.
Structure (Camb), 13, 75-85 Times cited: 21. [PubMed] [PDB]
Scholle MD, Collart FR, Kay BK (2004)
In vivo biotinylated proteins as targets for phage-display selection experiments.
Protein Expr Purif, 37, 243-52 Times cited: 6. [PubMed]
Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H,
Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM (2002)
Recognition of a virus-encoded ligand by a natural killer cell activation receptor.
Proc Natl Acad Sci U S A, 99, 8826-31 Times cited: 181. [PubMed]
Stevens FJ, Kuemmel C, Babnigg G, Collart FR (2004)
Efficient recognition of protein fold at low sequence identity by conservative application
of Psi-BLAST: application. J Mol Recognit, 18, 150-157 Times cited: 2. [PubMed]
Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI (2002)
A new vector for high-throughput, ligation-independent cloning encoding
a tobacco etch virus protease cleavage site. Protein Expr Purif, 25, 8-15 Times cited: 57. [PubMed]
Stols L, Millard CS, Dementieva I, Donnelly MI (2004)
Production of selenomethionine-labeled proteins in two-liter plastic bottles
for structure determination. J Struct Funct Genomics, 5, 95-102 Times cited: no data. [PubMed]
Stols L, Zhou M, Eschenfeldt WH, Millard CS, Abdullah J, Collart FR, Kim Y, Donnelly MI (2007)
New vectors for co-expression of proteins: Structure of Bacillus subtilis ScoAB obtained by
high-throughput protocols. Protein Expr Purif, 53, 396-403 Times cited: 0. [PubMed]
Yoon JR, Laible PD, Gu M, Scott HN, Collart FR (2002)
Express primer tool for high-throughput gene cloning and expression.
Biotechniques, 33, 1328-33 Times cited: 3. [PubMed]
...
For a more exhaustive list of publications see the MCSG publications website.
|