|
| |
 MCSG tries to automate the process of fold recognition, assignment and model validation, automate deposition of structures to databases, and speed up data release. Development a database to monitor and evaluate all steps of the process from gene to structure is essential. The database creates a self-training system to aid decision making process and improve efficiency. MCSG tries to automate the process of fold recognition, assignment and model validation, automate deposition of structures to databases, and speed up data release. Development a database to monitor and evaluate all steps of the process from gene to structure is essential. The database creates a self-training system to aid decision making process and improve efficiency. |
|
| |
 | Assessment of the methods of predicting function from structure | | Assessment of the methods of predicting function from structure |
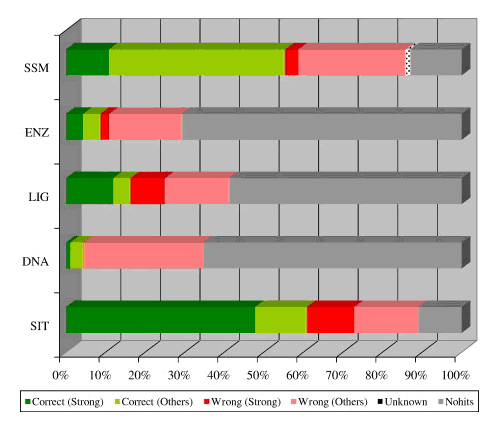
We have completed an assessment of the predictions made by the various structure-based methods incorporated in the ProFunc server. Our data set consisted of all MCSG structures of known function solved during PSI-1. The analysis suggested that the two most successful of the structure-based methods are the fold-matching (SSM) and reverse template (SIT) methods. These can occasionally identify function when sequence methods have failed, but typically strengthen or even confirm tentative functional assignments given by the sequence methods.
Manual assessment of the results proved to be a time-consuming process involving some subjective interpretation and, as a consequence, such a procedure is prone to human error. Thus, we developed a method based on a cut-down version of Gene Ontology (GO) schema, called GO-slims, to allow automated assessment of hits.
|
|
| |
| Improvements to the ProFunc server |
The ProFunc server returns results from many different methods. Often the different methods show a high level of agreement. To make such agreements more apparent a panel has been added to each results page to give a summary of commonly recurring terms in the names of the matched hits. For example, if many of the methods match to cupin, then the term “cupin” will appear often in the protein names. The panel scores each matched term according to the confidence of the prediction and these scores are accumulated and summarized. A second panel compiles the most commonly occurring terms from the GO assignments associated with the matched sequences/structures.
|
|
| |
| Improvements to the ProFunc server |
We have recently set up three additional services for use by the MCSG collaborators:
- An MCSG priority upload server (http://www.ebi.ac.uk/thornton-srv/databases/profunc/mcsg_index.html). This allows structures of particular interest to be uploaded to the ProFunc server and the results copied to us once done. This allows us to examine the ProFunc analyses and alert the depositors to any noteworthy results that may be of use when the structure is published.
- A reverse templates server called "Tempura" (http://www.ebi.ac.uk/thornton-srv/databases/tempura). This server is specifically for running only the reverse template analysis from ProFunc, but with some scope for the user to guide the searches by specifying particular regions of the protein to focus on. The user uploads a coordinate file and is then presented with a list of residues to pick from. Templates are then generated from only the specified residues and the template search is run as normal. Additionally, the user can specify which structures to run against, thus allowing comparisons of active sites within family members or simply between pairs of proteins. This server is still in development and will be published later this year.
- A wiki site (http://www.ebi.ac.uk/thornton-srv/databases/mcsgwiki) has been set up for use by the MCSG consortium and contains an entry for every MCSG structure. Each entry is automatically created when we receive the coordinate file from the MCSG database. The ProFunc results are automatically transferred into wiki format and put on this site. The aim of this service is to allow for collaborators across the sites to comment on the functional predictions for each protein and to share the results of experimental validations (or failed tests). Particular hits can be annotated as being of special importance and any MCSG member can add their comments and initiate an online discussion.
|
|
| |
| Cognate ligands |
As a first stage to identifying which ligands might bind to a given protein structure we have completed the PROCOGNATE database (http://www.ebi.ac.uk/thornton-srv/databases/procognate) which provides a mapping between cognate ligands and protein structural domains (Bashton et al., 2006). The database pulls together data from MSD, KEGG, CATH, SCOP and UniProt and can be searched in a variety of ways, including PDB code, PDB ligand, EC number, KEGG reaction, KEGG compound, SCOP and CATH superfamilies and free-text searches on ligand and structure names. So far we have completed the back-end database and are currently working on a prototype website front-end.
|
|
| |
| Homology modeling based on MCSG structures – The GeMMA web site |
GeMMA is the Genome Modeling and Model Annotation website (http://www.biochem.ucl.ac.uk/~dlee/GeMMA/) providing access to homology models based on MCSG solved structures together with annotations and derived data. An improvement made to the original GeMMA site in the last year is the generation and annotation of clusters of close homologues for each homology model and template (MCSG target structure). Software has been written to generate multiple sequence alignments with columns colored by the level of conservation calculated by Scorecons. These colors are mapped onto the surface of the relevant structure in a RasMol display. Work is underway on a scoring scheme to compare conserved residues between different clusters so that relatively distantly related models with a high probability of being closely functionally related can be linked together.
|
|
| |
|
The Global Protein Surface Server (GPSS) |
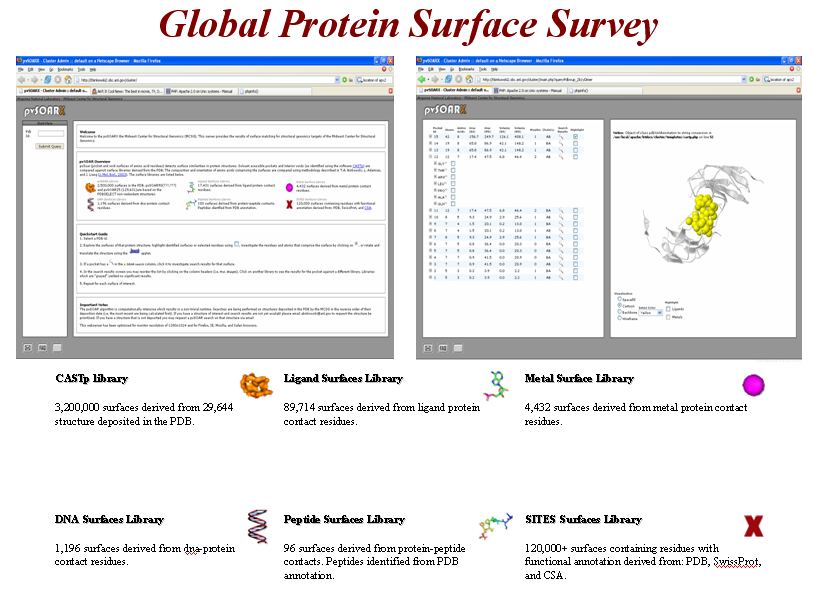
Uncovering similar surface patterns between proteins has been useful for providing insightful ideas about biochemical functions and mechanisms of active sites. In cases of low backbone sequence identity or low overall structural similarity, protein surface analysis has proven to be a powerful new method for identifying distant relationships between proteins. Surface analysis is a subset of the larger field of structural template based protein analysis wherein functional is transferred based on similarity to an annotated template. The Global Protein Surface Survey (GPSS) provides a comprehensive library of annotated surface templates from all structures deposited in the PDB, which are being used to identify functions of structural genomics proteins.

Annotated 3D Surface Libraries.
Automated identification and calculation of functionally annotated surfaces is accomplished through two methods: mapping of functionally annotated residues from public sources onto a CASTp surface or through exclusion contact surface identification for ligand, metal, DNA, and peptide binding sites. All functionally annotated surfaces are organized into a GPSS web server, which makes the data publicly available through a web interface or through a plugin to the PyMOL molecular visualization program (http://gpss.mcsg.anl.gov). During the 2006 calendar year, there have been 83,619 unique surface requests to the server. The GPSS PyMOL plugin has been downloaded 2,374 times.
Protein Surface Comparison.
A new methodology, SurfaceAlign, has been developed to extend our research into annotating newly determined proteins with unknown function. Utilizing global shape and local physicochemical texture, we shape match protein surfaces against libraries of annotated surfaces. Several comparison metrics (inter-atomic distance distributions, spatial conservation of amino acids and volume overlap of aligned structures) evaluate the complementarity between surfaces.
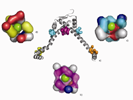
SurfaceAlign has proven useful in identifying distant relationships between proteins with no sequence or no structural homology. It has also allowed us to identify evidence of convergent evolutions, a challenging task for functional annotation. This is shown for two structural genomics targets IsdG and IsdI from S. aureus 6 (PDB ids = 1xbw and 1sqe). The IsdG and IsdI proteins show no significant sequence similarity to known heme-monooxygenases and do not contain the conserved N-terminal histidine or the GXXXG motif characteristic for these enzymes, yet display classical heme-monooxygenase activity. Also, while all known members of heme-oxygenase superfamily are of all α-helical fold, both IsdG and IsdI adopt α+β sandwich with an anti-parallel β-sheet and ferredoxin-like fold and form a β-barrel at the dimer interface.
The structure of IsdG has a prominent pocket (surface area=332.7Ǻ2, volume=157.7Ǻ3) formed between α-helices and beta sheets (Fig. E14, yellow). Querying this surface against the GPSS ligand surface library reveals a striking similarity to the heme binding pocket in heme oxygenase (HMUO) from C. diphtheriae (PDB id=1iw0, surface area= 427.7Ǻ2, volume=232.5Ǻ3) (Figure E14, green). There are 19 conserved residues between the surfaces that exhibit statistically significant similarity. The surface alignment is shown with the ligand from HMUO. Similar results are obtained for IsdI.
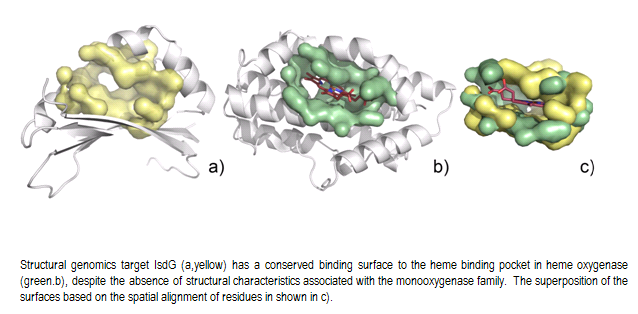
|
|
| |
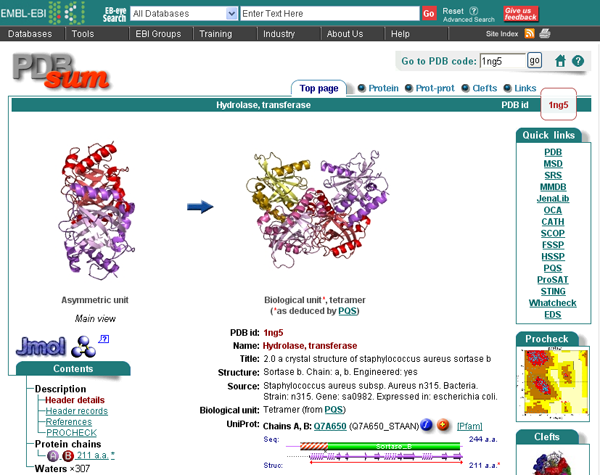
| PDBsum |
The PDBsum is a pictorial database that provides an at-a-glance overview of the contents of each 3D structure deposited in the Protein Data Bank. It shows the molecule(s) that make up the structure (ie protein chains, DNA, ligands and metal ions) and schematic diagrams of their interactions. Extensive use is made of the freely available RasMol molecular graphics program to view the molecules and their interactions in 3D.
The information given on each PDBsum entry is spread across several pages, as listed below and accessible from the tabs at the top of the page. Only the relevant tabs will be present on any given page.
Top page - summary information including thumbnail image of structure, molecules in structure, enzyme reaction diagram (where relevant), GO functional assignments, and selected figures from key reference
Protein - wiring diagram, topology diagram(s) by CATH domain, and residue conservation (where available)
DNA/RNA - DNA/RNA sequence and NUCPLOT showing interactions made with protein
Ligands - description of bound molecule and LIGPLOT showing interactions made with protein
Prot-prot - schematic diagrams of any protein-protein interfaces and the residue-residue interactions made across them
Clefts - listing of top ten clefts in the surface of the protein, listed by volume with any bound ligands shown
Links - links to external databases
|
|
| |
 Selected related publications: Selected related publications:
Binkowski TA, Joachimiak A, Liang J (2005)
Protein surface analysis for function annotation in high-throughput structural genomics pipeline.
Protein Sci, 14, 2972-81 Times cited: 3. [PubMed]
Laskowski RA (2001)
PDBsum: summaries and analyses of PDB structures.
Nucleic Acids Res, 29, 221-2 Times cited: 91. [PubMed]
Laskowski RA, Watson JD, Thornton JM (2005)
ProFunc: a server for predicting protein function from 3D structure.
Nucleic Acids Res, 33, W89-93 Times cited: 28. [PubMed]
Watson JD, Sanderson S, Ezersky A, Savchenko A, Edwards A, Orengo C, Joachimiak A, Laskowski RA, Thornton JM (2007)
Towards Fully Automated Structure-based Function Prediction in Structural Genomics: A Case Study.
J Mol Biol, , Times cited: 0. [PubMed]
Zhang R, Wu R, Joachimiak G, Mazmanian SK, Missiakas DM, Gornicki P, Schneewind O, Joachimiak A (2004)
Structures of Sortase B from Staphylococcus aureus and Bacillus anthracis Reveal Catalytic Amino Acid Triad in the Active Site.
Structure (Camb), 12, 1147-56 Times cited: 10. [PubMed] [PDB]
...
For a more exhaustive list of publications see the MCSG publications website.
|
 MCSG tries to automate the process of fold recognition, assignment and model validation, automate deposition of structures to databases, and speed up data release. Development a database to monitor and evaluate all steps of the process from gene to structure is essential. The database creates a self-training system to aid decision making process and improve efficiency.
MCSG tries to automate the process of fold recognition, assignment and model validation, automate deposition of structures to databases, and speed up data release. Development a database to monitor and evaluate all steps of the process from gene to structure is essential. The database creates a self-training system to aid decision making process and improve efficiency.