|
| |
 A critical issue in structural genomics, and in structural biology in general, is the availability of high-qualitysamples. The additional challenge in structural genomics is the need to produce high numbers of proteins withlow sequence similarities and poorly characterized or unknown properties. ‘Structural-biology-grade’ proteinsmust be generated in a quantity and quality suitable for structure determination experiments using X-ray crystallographyor nuclear magnetic resonance (NMR). The choice of protein purification and handling proceduresplays a critical role in obtaining high-quality protein samples. The purification procedure must yield a homogeneousprotein and must be highly reproducible in order to supply milligram quantities of protein and/or itsderivative containing marker atom(s). At the Midwest Center for Structural Genomics we have developed protocolsfor high-throughput protein purification. These protocols have been implemented on AKTA EXPLORER3D and AKTA FPLC 3D workstations capable of performing multidimensional chromatography. The automatedchromatography has been successfully applied to many soluble proteins of microbial origin. A critical issue in structural genomics, and in structural biology in general, is the availability of high-qualitysamples. The additional challenge in structural genomics is the need to produce high numbers of proteins withlow sequence similarities and poorly characterized or unknown properties. ‘Structural-biology-grade’ proteinsmust be generated in a quantity and quality suitable for structure determination experiments using X-ray crystallographyor nuclear magnetic resonance (NMR). The choice of protein purification and handling proceduresplays a critical role in obtaining high-quality protein samples. The purification procedure must yield a homogeneousprotein and must be highly reproducible in order to supply milligram quantities of protein and/or itsderivative containing marker atom(s). At the Midwest Center for Structural Genomics we have developed protocolsfor high-throughput protein purification. These protocols have been implemented on AKTA EXPLORER3D and AKTA FPLC 3D workstations capable of performing multidimensional chromatography. The automatedchromatography has been successfully applied to many soluble proteins of microbial origin.
One of the main objectives of the PSI pilot projects were to develop technologies for production of proteins in milligram quantities reliably, reproducibly, quickly, and at low cost. For crystallography applications the resulting protein samples must be compatible with the crystallization process. The protein in the sample must be folded and soluble, as well as chemically, conformationally, and functionally homogeneous. The sample must be free of critical contaminants that may degrade, denature, destabilize, or modify protein or interfere with crystallization or structure determination. Protein purity of >95% is typically required. Protein samples must be stable during crystallization trials, suitable for incorporation of heavy atoms to aid structure determination, and functionally relevant. The quantities of proteins in the samples must allow achieving protein concentrations in the range of 5–25 mg/ml, testing 200–500 crystallization conditions, growing X-ray-quality single crystals, establishing cryoconditions, and producing rational heavy atom derivatives for structure determination.
We have developed standard operating procedures for protein purification that make protein samples suitable for automated structure determination using synchrotron-based X-ray crystallography. These standard operating procedures are based on the following principles:
- All proteins are expressed as a fusion with a uniform, cleavable affinity tag and protected against proteolysis with several protease inhibitors.
- Proteins are purified using affinity chromatography followed by buffer-exchange chromatography, to promote protein solubility and efficient tag removal.
- The affinity tag is cleaved off by a specific tagged protease.
- The protein is further purified using affinity chromatography followed by buffer-exchange chromatography compatible with protein concentration and crystallization methods.
 |
|
| |
 | IMAC-I and buffer exchange steps | | IMAC-I and buffer exchange steps |
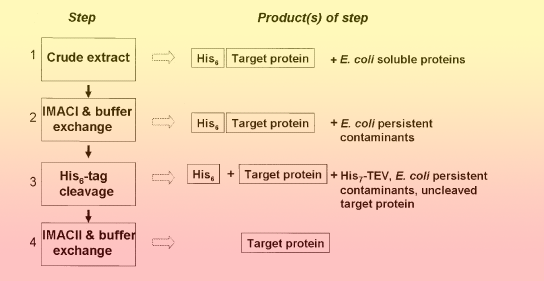
All chromatography experiments were performed at 4 ºC. Crude extracts of six proteins (typically 15–50 mL) were applied by the sample pump (flow rate 1 mL/min) sequentially onto six 5-mL HiTrap chelating HP columns charged with Ni +2. The columns were washed with 10 column volumes (CV) of buffer A, followed by 15 CV of buffer A containing 20 mM imidazole (flow rate 5 mL/min). Each protein was first eluted to a 10-mL loop with buffer A containing 250 mM imidazole (flow rate 2 mL/min), then applied to a HiPrep 26/10 desalting colum pre-equilibrated with buffer A. Just prior to injecting protein onto the desalting column, 2 mL of 5 mM EDTA in buffer A was injected onto the desalting column to create a slow-moving EDTA zone on the desalting column and sequester any Ni +2 ions released from the chelating column. The buffer exchange step was run at a flow rate of 8 ml/min. The desalting column was washed and re-equilibrated prior to the next purification cycle. The tubing and loop were washed between chromatography steps to avoid cross-contamination. The final peak fractions and all solutions that could contain target protein were collected.
Throughout the purification process, several parameters, including UV absorbance, pressure, flow rate, pH, and ionic strength, were monitored and logged (Figure 2). All fractions were analyzed and documented and all data stored in a single results file. The purification processes in this experiment took 12–15 h for six proteins, depending on the initial sample volumes. The chelating columns were recycled four to five times using an automated procedure by metal stripping with 50 mM EDTA and charging with 100 mM NiSO4. |
|
| |
| IMAC-II and buffer-exchange steps |
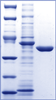
Proteins purified with IMAC-I and buffer exchange were treated with the His7-tagged TEV protease to remove the His6-tag for 16–24 h at 4 ºC following the basic protocol (see above). Cleavage was monitored by SDS-PAGE and Coomassie Brilliant Blue R (Amersham Biosciences) staining. After the cleavage, the reaction mixture containing target protein (cleaved and some uncleaved), His7-tagged-TEV protease and His6-tag was applied to a 1-ml chelating affinity column and the column was washed with 3 CV of buffer A. All chromatographic steps were performed at 22 ºC. The column flow-through and wash fraction was first collected onto a 20-mL loop and then applied to a customized desalting column Sephadex G-25 fine XK 26/20 equilibrated with storage buffer containing 20 mM Tris/HCl 7.5, 500 mM NaCl, and 2 mM DTT. Protein was eluted with storage buffer, protein peaks were collected in 2-mL fractions (Figure 3) and analyzed by the SDS-PAGE stained with Coomassie Brilliant Blue R (Figure 4). Purification of six proteins takes about 9 h. The 1-mL chelating columns were recycled four to five times using the automated procedure described above. |
|
| |
| Protein characterization |
Protein Characterization |
| Purity |
SDS-PAGE stained with Coomassie Brilliant Blue and lab-on-the-chip 2100 Bioanalyzer (Agilent) |
| Concentration |
Coomassie Plus Protein Assay (Pierce, Catalog No. 23236) and UV spectrometry |
| Poly-dispersity |
Dynamic light scattering (DynaPro, Protein Solutions) |
| Estimated molecular weight in solution |
Size exclusion chromatography |
| Suspected chemical heterogeneity and bound ligands |
Mass spectrometry (nLC-ESI MS/MS and MALDI-TOF; QSTAR-XL) |
| Bound ligands |
UV/Vis spectrometry |
|
|
| |
| Protein concentration and storage |
All proteins were concentrated with Centricon Plus Centrifugal Filter Units (Millipore), using molecular weight cutoff as recommended by the manufacturer. All proteins were flash frozen in 50 μL aliquots in liquid nitrogen temperature in the storage buffer and stored in an LS6000 liquid nitrogen storage system (Taylor-Wharton) for an extended period of time.
|
|
| |
| Platform for automated multidimensional chromatography |
MCSG collaborated with Amersham Biosciences to adapt the AKTA Explorer 100A for multidimensional automated chromatography needs. The complete AKTA EXPLORER 3D system consists of the AKTA Explorer 100A, UNICORN software version 4.0 or higher, sample pump P-950, fraction collector Frac- 950, a 3D kit that allows attachment of multiple columns and loops, multi-channel UV/Vis spectrometer, two sample loops, and an air sensor. The AKTA EXPLORER 3D system executes a series of commands to perform automated purification and sample collection. The sample pump includes an in-line air sensor that allows unattended direct loading of crude protein extracts. The system is capable of purifying up to seven protein samples. Samples are loaded serially on (1) up to seven singlestep IMAC columns, (2) up to six IMAC columns, each followed by a buffer-exchange chromatography step, or (3) up to five IMAC columns, followed by buffer-exchange chromatography and another chromatographic step. In between chromatographic steps, the samples are stored in sample loops (10 mL and 20 mL). Automated peak detection allows collection of target protein peaks and other relevant fractions into appropriate loops or in the fraction collector. AKTA FPLC is similarly outfitted for multidimensional chromatography (AKTA FPLC 3D). Using UNICORN software, we have developed several methods for automated protein purification, as well as for automated charging of chelating columns that utilize the chemistry of metal stripping followed by recharging of the matrix with Ni +2. Potential problems with leaching of Ni +2 during purification have been addressed |
|
| |
| On column cleavage |
Our purification pipeline follows two Ni-IMAC steps which include IMAC-I, tag-cleavage using His-tagged TEV protease, and IMAC-II. This whole process can take 5 to 6 days to accomplish, including 2 - 3 days of tag-cleavage. In order to shorten the process, we have developed an on-column tag cleavage approach in which the entire purification procedure is done on the AKTAxpress chromatography workstation without manual intervention. After a target protein with an affinity tag is applied to an affinity column and washed, a tagged TEV protease is then applied to the same column and incubated for 16 hours at 30°C. The target protein without the tag is then eluted while the tag, un-cleaved protein, and the protease remain on the column. The temperature and incubation time are controlled by the software and electronics.
|
|
| |
| Protein reductive methylation – progress towards automation |
We implemented the reductive methylation of lysine residues (Rayment et. al., Volume 276 of Methods of Enzymology, 1997, 276:171-9) for proteins that either failed to crystallize or initially produced poor-diffracting crystals. Thus far, the reductive methylation approach has been applied to more than 300 proteins, which produced more than 100 crystals, and 23 structures. Semi-automated methylation procedures using the Biomek® FX Workstation have been developed that reduce the process time from ~ 20 hours to 2 – 4 hours. We are in the process of integrating this automated process into our high-throughput protein purification pipeline, which now includes an automated, “on-column tag cleavage” via AKTAxpress™.
 |
|
| |
| Work on salvaging projects with poor or no crystals |
We have developed a number of salvaging approaches for proteins that fail in the initial MCSG structure determination pipeline (MCSG SDP). UT SWMC focuses on some of the problems that involve major crystallography difficulties.
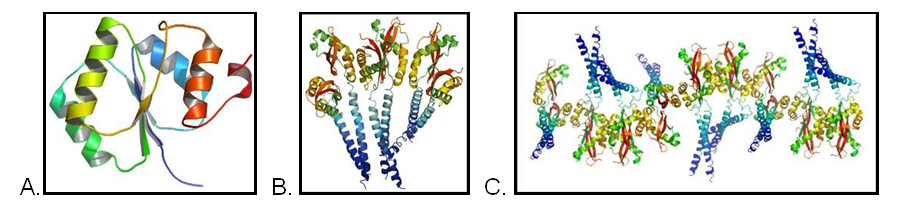
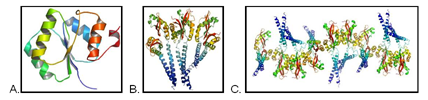
The diffraction data for three structures (A, B, C) was collected some time ago but were refractory to the high-throughput approach. Detailed analysis of the projects revealed very complex non-crystallographic symmetry (A) and very large anisotropic motions (B and C).
 |
|
| |
 Selected related publications: Selected related publications:
Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A (2004) Automation of protein purification for structural genomics. J Struct Funct Genomics, 5, 111-8 Times cited: no data. [PubMed]
...
For a more exhaustive list of publications see the MCSG publications website.
|
|