 The objective of the Midwest Center for Structural Genomics (MCSG) is to develop and optimize new, rapid, integrated methods for highly cost-effective determination of protein structures through X-ray crystallography using a third-generation synchrotron. Some of the goals are to develop standard multiwavelength anomalous diffraction (MAD) protocols and optimize anomalous dispersion strategies for rapid structure determination using synchrotron radiation, increase the speed of structure determination using parallel approaches, and minimize manual intervention in model building and structure refinement. The objective of the Midwest Center for Structural Genomics (MCSG) is to develop and optimize new, rapid, integrated methods for highly cost-effective determination of protein structures through X-ray crystallography using a third-generation synchrotron. Some of the goals are to develop standard multiwavelength anomalous diffraction (MAD) protocols and optimize anomalous dispersion strategies for rapid structure determination using synchrotron radiation, increase the speed of structure determination using parallel approaches, and minimize manual intervention in model building and structure refinement. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Modern synchrotron protein crystallography facilities provide tunable and stable sources of x-rays in the 6-20 keV range. There are numerous advantages to using dedicated synchrotron beamlines for protein crystallography:
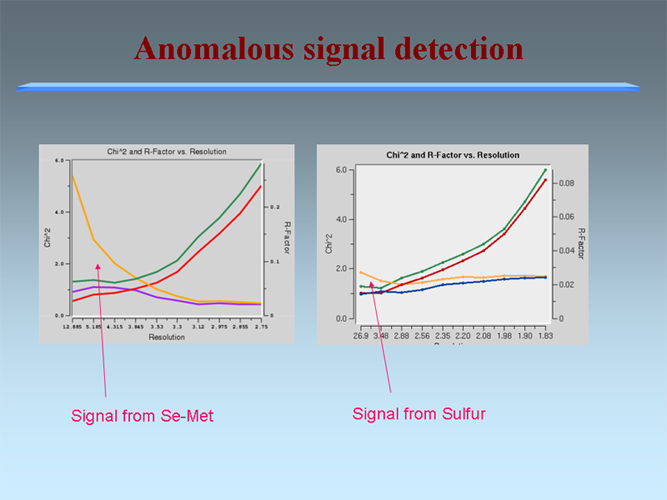
For alFor all proteins containing methionine, we produce SeMet derivative. This approach has proven to be the most cost effective (>94% of MCSG structures have been phased with selenium atoms). For proteins that fail we consider alternative phasing strategies (other rational derivatives such as Br and Xe and co-crystallization with metals (Rb, Zn, Hg, lanthanides)). Our results show that an anomalous diffraction signal from a single SeMet residue is usually sufficient for structure determination. The MCSG pipeline allows a structure to be solved from one wavelength during data collection for a second wavelength. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
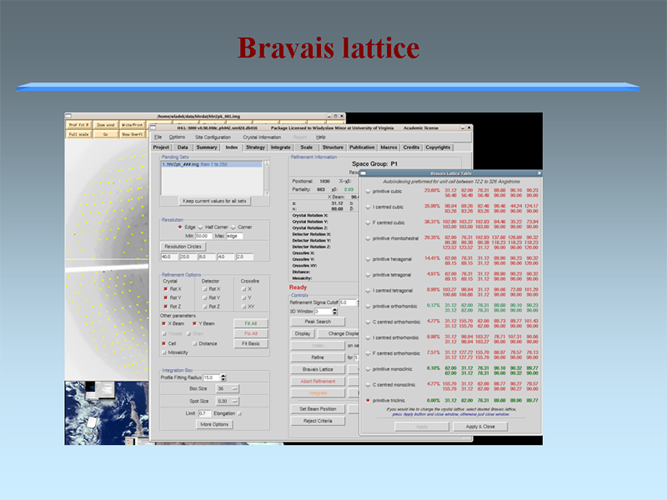
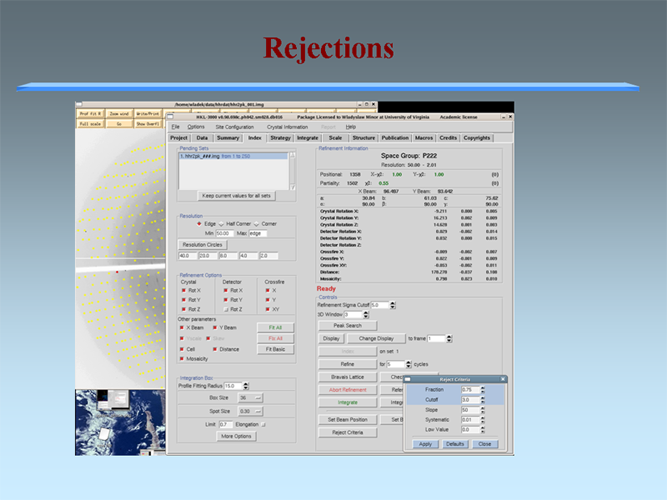
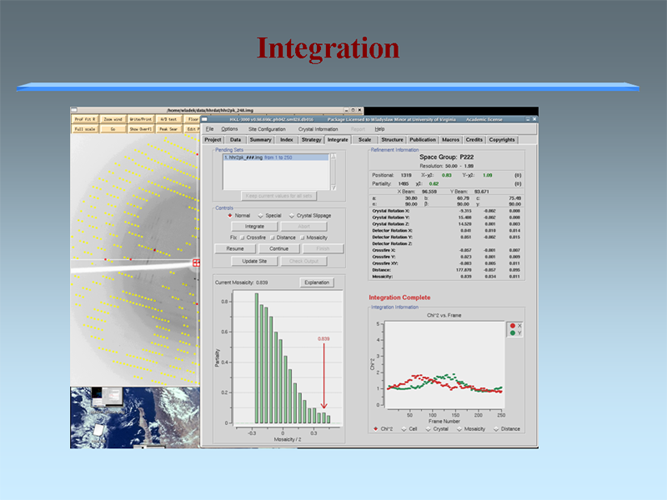
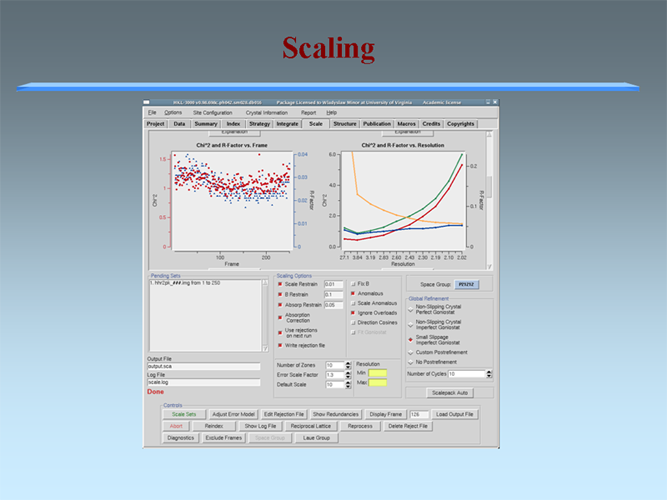
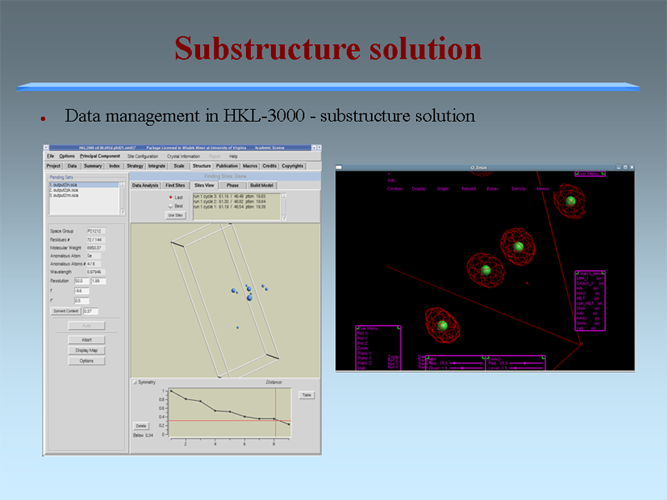
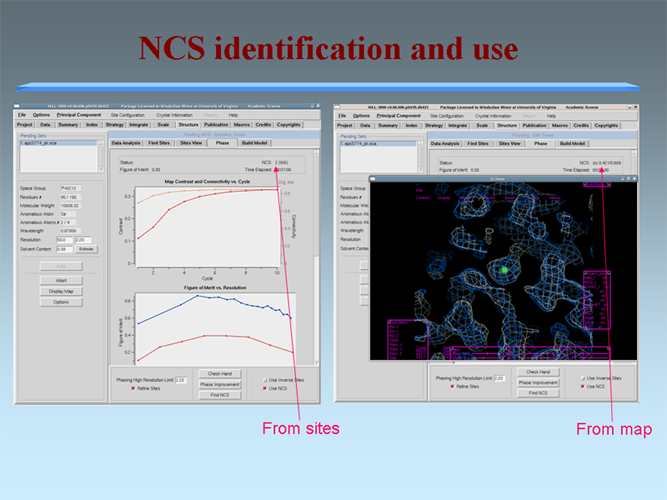
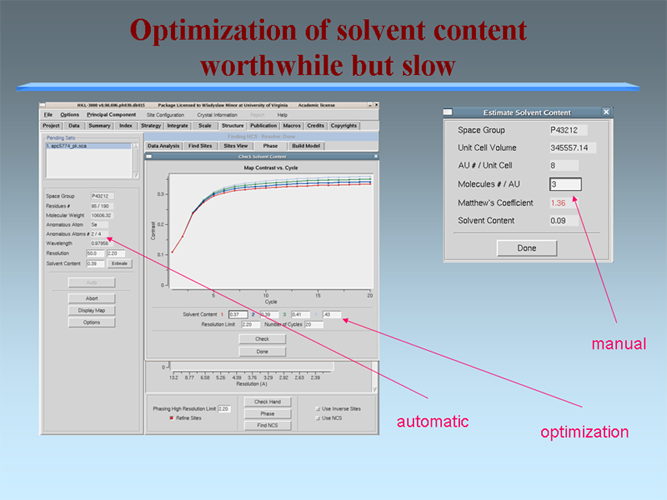
The MCSG has developed semi-automated approaches to structure determination that is implemented in HKL3000 package. Our goal is to provide the experimenter with near-real-time feedback by integrating the x-ray diffraction experiment with a rapid evaluation of data quality and structure solution. This approach has already improved the success rate of x-ray diffraction experiments at the synchrotron beamlines and significantly increased the efficiency of structure determination. We have built a system to analyze data and carry out the various computational steps needed for crystallographic structure determination using alternative data-processing paths. The HKL3000 integrates data collection, reduction, phasing, and model building — significantly accelerating structure determination and reducing the number of data sets required for a single structure determination. This system makes parallel attempts to solve the structure using different algorithms and exploring different approaches and is combined with relational databases and linked to external web resources. This HKL3000 converts diffraction data to an interpretable electron density map, and for smaller structures, into an initial model. Automated model building is done using the SOLVE/RESOLVE and ARP/wARP program suites. The system is interfaced with MCSG’s SGPDB and with PDB, Swissprot, and other generally available databases. The current version of our semi-automatic system has solved over 300 new structures. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
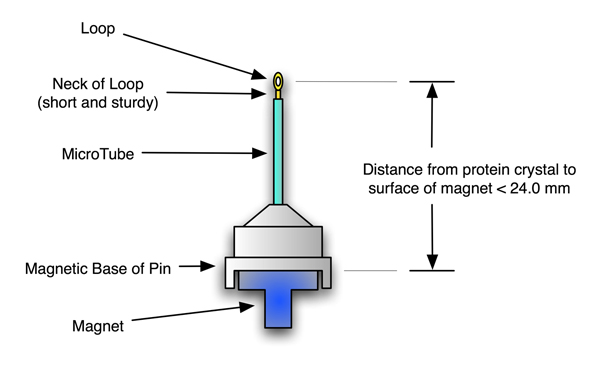
Cryo-pins we currently recomend for mounting on the Kappa head using magnetic mounts may be a maximum of 24.5 mm, magnetic base to protein crystal, inclusive.
Other sizes or styles or heights may be able to be accommodated, including
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A small workbench area will be available for use during data collection time. Due to the presence of other scheduled Users, access to the laboratory may only be available immediately before and after the assigned data collection time. A walk-in, +5° C cold room is available. One biochemistry workbench area is available in the cold room, and is shared by all 19ID and 19BM Users. One fume hood is present. Work involving either freezing of crystals in liquid-propane or heavy-atom compounds must be performed in this fume hood. Microscopes are available for viewing crystals. Two microscopes are available for 19ID; one in the biochemistry laboratory, one inside the beamline experimental hutch. Two additional, analogous microscopes are available for beamline 19BM. A fifth microscope is available in the cold room (+5° C). Preferential access to these microscopes is given to User groups actively collecting data at the beamlines. All microscopes are locked down in position, and may not be moved. Limited laboratory equipment is available for general use by all SBC Users. Although SBC staff try to maintain equipment in good working order, please be aware that heavy use may lead to repairs and unavailability of the equipment during your assigned beamtime. As always, if you feel the success of your experiment will depend upon the availability of consumable items, please bring your own supplies with you from your home institution.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Cryo-equipment available for users:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Where? How?
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In each beamline control area SBC also maintains a desktop PC running Windows XP Professional. These systems are used primarily as an option for performing data backup to Windows compatible firewire disks. A high speed Network Attached Storage device provides over 4 Terabytes of storage per beamline.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Borek D, Ginell SL, Cymborowski M, Minor W, Otwinowski Z.
Borek D, Minor W, Otwinowski Z (2003)
Korolev S, Dementieva I, Sanishvili R, Minor W, Otwinowski Z, Joachimiak A
(2001)
Minor W, Tomchick D, Otwinowski Z (2000)
Otwinowski Z, Borek D, Majewski W, Minor W (2003)
Walsh MA, Dementieva I, Evans G, Sanishvili R, Joachimiak A.
Walsh MA, Evans G, Sanishvili R, Dementieva I, Joachimiak A. MAD data collection - current trends.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}